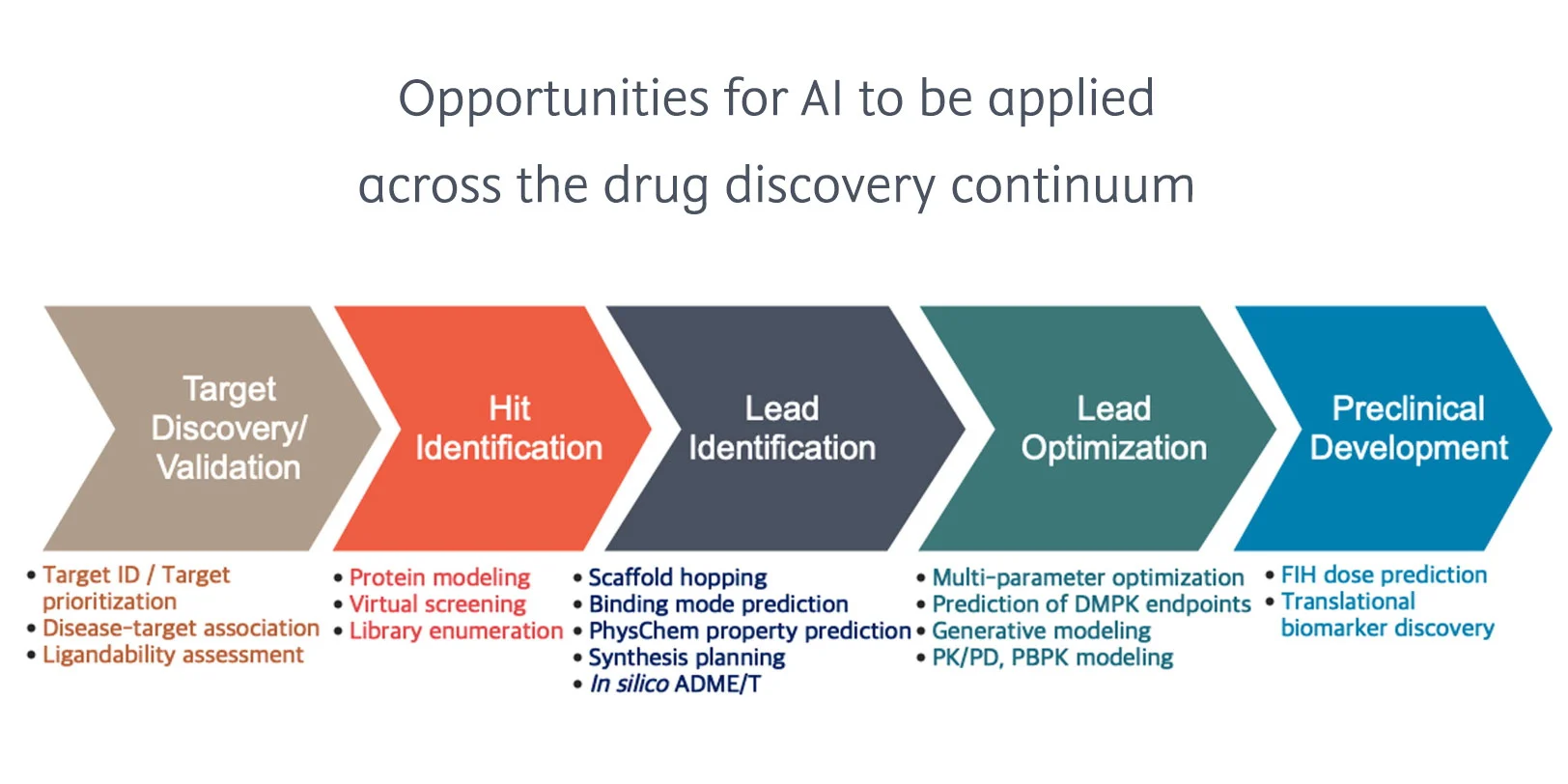
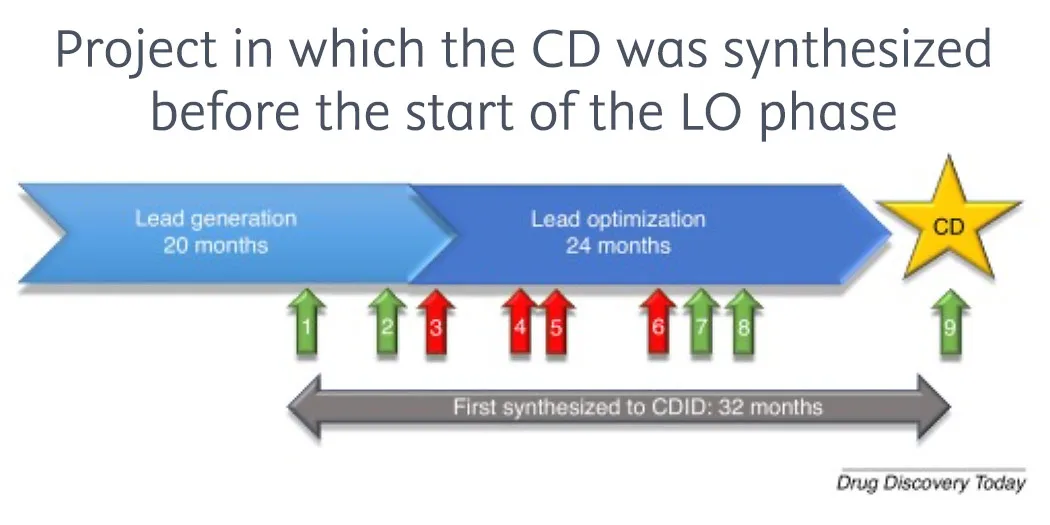
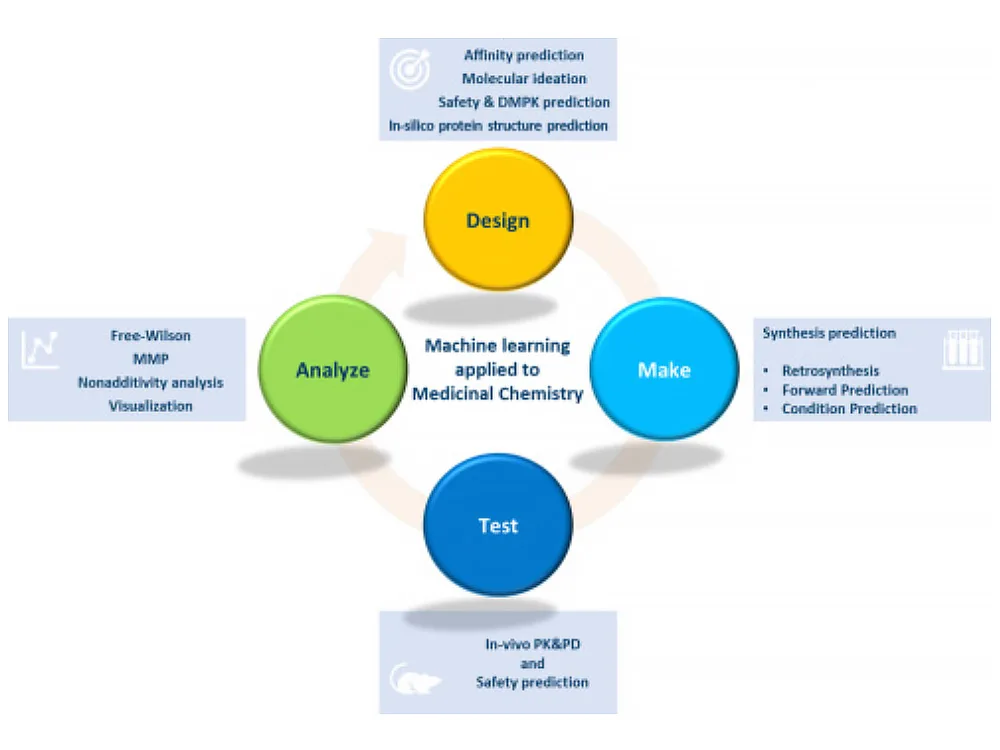
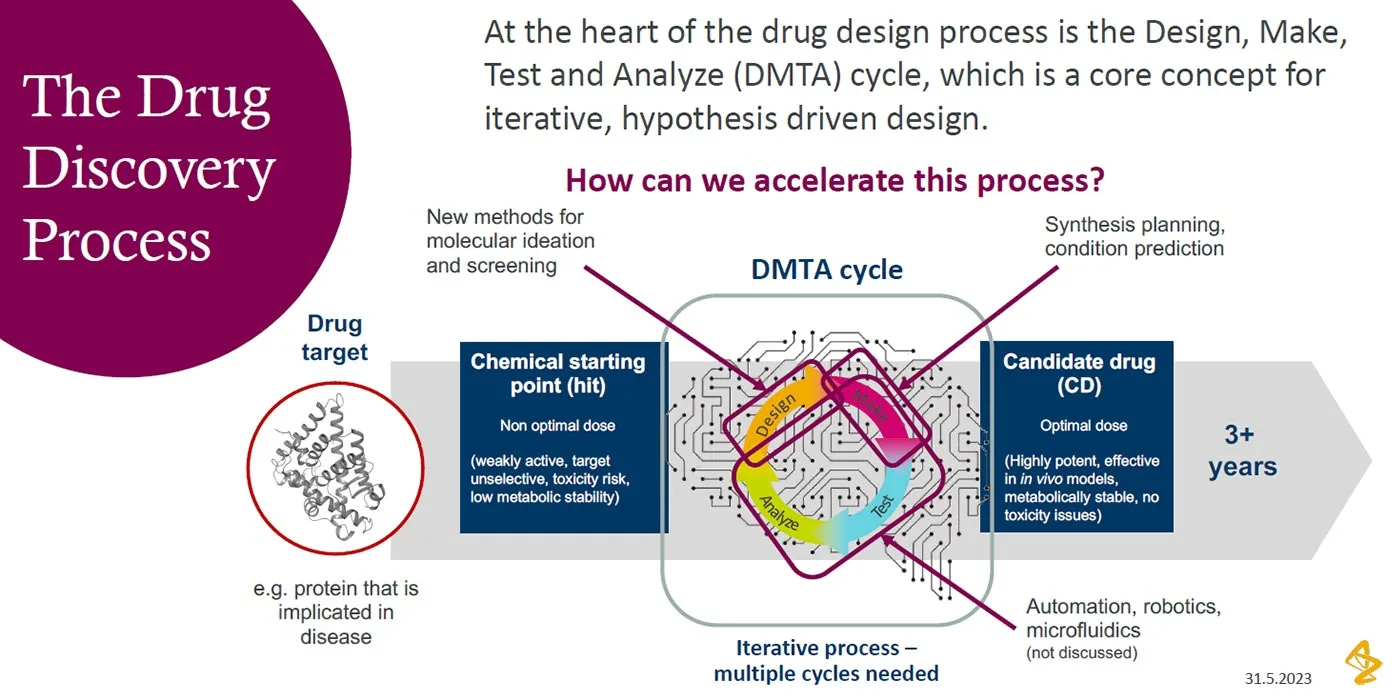
Checklist for successful predictive retrosynthesis
The following checklist can help chemists apply predictive retrosynthesis successfully:
☑ Do you understand the types/variety of data you need access to?
☑ Are you able to access all of these data sources both internally and externally?
☑ Are you confident data is trustworthy and high quality?
☑ Can you define the problem and prediction task clearly?
☑ How will you validate and determine the predictive performance of a model?
☑ Do you understand the model’s interface parameters so you can build a search that leads to desired results?
☑ Do you fully understand the structure and functionality of the target molecule?
☑ Have you identified any potential challenges associated with its synthesis?
☑ Have you decided parameters for selecting a synthesis route, including cost, yield, environmental impact?
☑ Have you identified partners and experts to collaborate with?